(Click on pictures to enlarge – click “back” to close)
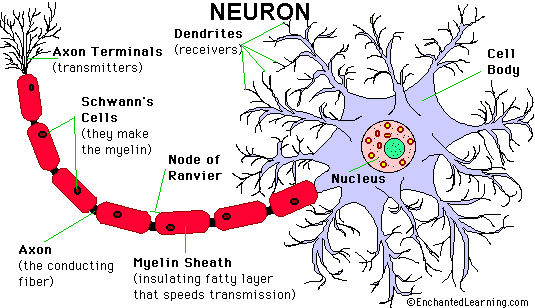
Peripheral nerves consist of fascicles that contain myelinated and unmyelinated axons. Endoneurium is the small amount of matrix that is present between individual axons. The perineurium is a sheath of special, fiber-like cells that ties the axons of each fascicle together. Epineurium is the connective tissue that surrounds the entire nerve trunk and gives off vascular connective tissue septa that traverse the nerve and separate fascicles from one another.
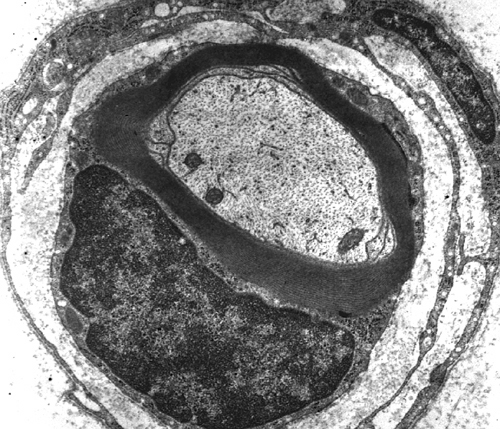 |  |
| Single myelinated axon | Normal nerve |
Axons thicker than one micron in the CNS and peripheral nervous system (PNS) are myelinated. Myelin is a spiral sheet of cell membrane wrapped around the axon. In the CNS, myelin is produced by oligodendroglial cells and in the PNS by Schwann cells. Each oligodendrocyte makes multiple segments of myelin that wrap around many axons. Each Schwann cell makes one segment of myelin. This is one reason why peripheral myelin regenerates more efficiently. Nodes of Ranvier are points of discontinuity between adjacent myelin sheaths in which the axon is not covered by myelin. Unmyelinated axons are covered by Schwann cell cytoplasm, but there is no spiraling of Schwann cell membrane around them.
The structure of central and peripheral myelin is essentially the same. Myelin is composed of 70% lipids and 30% protein. There are some important differences in myelin proteins between CNS and PNS. These differences explain why an allergic reaction against PNS myelin does not cause central demyelination and vice versa; and why inherited metabolic disorders of myelin proteins that affect peripheral nerves do not damage central myelin. On the other hand, lipids are similar between PNS and CNS myelin. For this reason, metabolic disorders of myelin lipids, such as metachromatic leukodystrophy, affect both, the central white matter and peripheral nerves.
The myelin sheath acts as an electrical insulator, preventing short-circuiting between axons. More important, it facilitates conduction. The nodes of Ranvier are the only points where the axon is uncovered by myelin and ions can be exchanged between it and the extracellular fluid. Depolarization of the axonal membrane at the nodes of Ranvier boosts the action potential that is transmitted along the axon and is the basis of saltatory (jumping) conduction.
The degree of regeneration and recovery depends on how well the cut ends are put together and on the extent of soft tissue injury and scarring around the area of transection. If reconstruction is not good, a haphazard proliferation of collagen, Schwann cell processes, and axonal sprouts fill the gap, forming a traumatic neuroma. Wallerian degeneration was initially described in experimental axotomy. Neuropathies characterized by Wallerian degeneration include those that are caused by trauma, infarction of peripheral nerve (diabetic mononeuropathy, vasculitis) and neoplastic infiltration.
In distal axonopathy, degeneration of axon and myelin develops first in the most distal parts of the axon and, if the abnormality persists, the axon “dies back”. This causes a characteristic distal (“stocking-glove”) sensory loss and weakness. Neurofilaments and organelles accumulate in the degenerating axon (probably due to stagnation of axoplasmic flow). Eventually the axon becomes atrophic and breaks down. Severe distal axonopathy resembles Wallerian degeneration. At an advanced stage, there is loss of myelinated axons. Many clinically important neuropathies caused by drugs and industrial poisons such as pesticides, acrylamide, organic phosphates, and industrial solvents are characterized by distal axonopathy.
Distal axonopathy is thought to be caused by pathology of the neuronal body resulting in its inability to keep up with the metabolic demands of the axon. This explains why the disease begins in the most distal parts of nerves, and large axons that have the highest metabolic and nutritional demands are more severely affected. However, this question is not settled. It is hard to imagine how the relatively miniscule neuronal body can keep up with the metabolic demands of the enormous mass of the axon. Furthermore, the neuronal body is just as dependent on the distal axon and its synapses for trophic interactions that keep it alive and functioning.
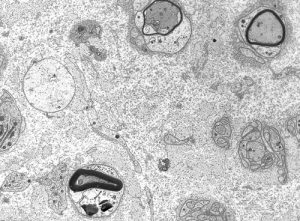
Segmental demyelination, originally described in experimental lead poisoning, is characterized by breakdown and loss of myelin over a few segments. The axon remains intact and there is no change in the neuronal body. The loss of saltatory conduction that results from segmental demyelination leads to decrease of conduction velocity and conduction block. Deficits develop rapidly but are reversible because Schwann cells make new myelin. However, in many cases, demyelination leads to loss of axons and permanent deficits. The nerve, in segmental demyelination, shows demyelinated axons, thin-regenerating-myelin, “onion bulbs”(see below) and, in severe cases, loss of axons. The status of myelin can be evaluated with teased fiber preparations of peripheral nerves and by electron microscopy. Neuropathies characterized by segmental demyelination include acute and chronic inflammatory demyelinative neuropathies, diphtheritic neuropathy, metachromatic leukodystrophy and Charcot-Marie-Tooth disease.
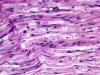
“Onion bulb” formations are concentric layers of Schwann cell processes and collagen around an axon. This proliferation is caused by repetitive segmental demyelination and regeneration of myelin and can cause gross thickening of peripheral nerves (hypertrophic neuropathy). The central axon is often demyelinated or has a thin layer of myelin. Onion bulb formations are the histological hallmark of Charcot-Marie-Tooth disease, but are also seen in other hereditary neuropathies (Dejerine-Sottas disease, Refsum disease), in diabetic neuropathy, and in chronic inflammatory demyelinative neuropathy.
The pathology of peripheral neuropathy is reflected in the spinal cord. Acute axonal neuropathy causes cental chromatolysis. Axonal neuropathy and distal axonopathy involving the bipolar neurons of the dorsal root ganglia cause degeneration of the central axons of these neurons in the gracile and cuneate tracts of the spinal cord. This lesion is associated with loss of position and vibration sense and sensory ataxia. Neuropathies can be classified on the basis of their pathological changes into axonal (Wallerian degeneration and distal axonopathy), demyelinative, or mixed.
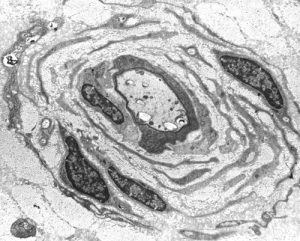
Hypertrophic neuropathy
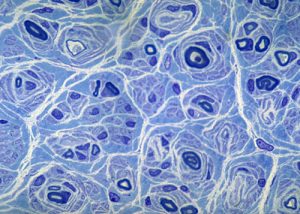
Hypertrophic neuropathy
The pathology of peripheral neuropathy is reflected in the spinal cord. Acute axonal neuropathy causes cental chromatolysis. Axonal neuropathy and distal axonopathy involving the bipolar neurons of the dorsal root ganglia cause degeneration of the central axons of these neurons in the gracile and cuneate tracts of the spinal cord. This lesion is associated with loss of position and vibration sense and sensory ataxia. Neuropathies can be classified on the basis of their pathological changes into axonal (Wallerian degeneration and distal axonopathy), demyelinative, or mixed.
The goal of the investigation of peripheral neuropathy is to establish the diagnosis of peripheral neuropathy, determine if it is an axonal or demyelinative process, and find its cause.
Clinically, neuropathy causes weakness and atrophy of muscle, loss of sensation or altered sensation (pain, paresthesias), and weak or absent tendon reflexes. Nerve conduction studies can distinguish demyelinative neuropathy (slowing of conduction velocity or conduction block) from axonal neuropathy (low-action potential amplitudes). Electromyography (EMG) can distinguish denervation atrophy from primary muscle disease. CSF examination is helpful, especially in inflammatory demyelinative neuropathies. Because cranial and spinal roots bathe in CSF, demyelinative neuropathies that involve roots cause elevation of CSF protein. Also, inflammation in nerve roots causes CSF pleocytosis. Careful history taking with attention to family history, environmental exposure, and systemic illness, combined with neurological examination and laboratory studies can determine the etiology in most peripheral neuropathies. When the diagnosis is in doubt, a nerve biopsy studied by light microscopy, electron microscopy, morphometry, and teased fiber preparations can give more definitive information. The sural nerve is usually chosen for biopsy because it is superficial and easy to find and it is predominantly sensory. Sural nerve biopsy leaves a patch of hypesthesia in the lateral aspect of the foot that is usually well tolerated.
Diabetic and other neuropathies affect predominantly small myelinated and unmyelinated fibers that convey pain and temperature sensation. Degeneration in these “small fiber neuropathies” involves the most distal portions of nerve fibers that are found in different organs and tissues (somatic fibers) rather than fibers in major nerves. Nerve conduction studies and EMG in such cases may be normal and the sural nerve biopsy may be difficult to interpret. The diagnosis can be made with a skin biopsy. A 3-4 mm plug of skin is removed with a punch and sectioned with a microtome. The sections are treated with antibodies to Protein Gene Product 9.5 which reveal small nerve fibers that penetrate the epidermis. The density of these fibers is reduced in small fiber neuropathies.
End stage axonal neuropathy
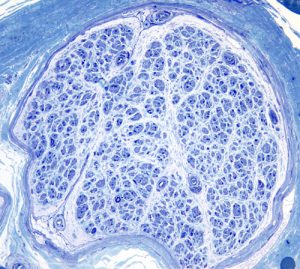
The pathological changes of most peripheral neuropathies (axonal degeneration, segmental demyelination or a combination of these) are not specific. In any active neuropathy, there are macrophages removing myelin and axon debris. Advanced axonal neuropathy shows loss of myelinated axons and increased endoneurial collagen. Some chronic demyelinative neuropathies show hypertrophic changes. Thus, in most neuropathies, the sural nerve biopsy can only establish the diagnosis of neuropathy and distinguish axonal from demyelinative and acute from chronic neuropathy, but cannot determine the cause of neuropathy. Only a few peripheral neuropathies show disease-specific pathological changes allowing a specific diagnosis. These neuropathies include acute and chronic inflammatory demyelinative neuropathies, hereditary motor and sensory neuropathies, vasculitis, sarcoid neuropathy, leprosy, amyloid neuropathy, neoplastic invasion of peripheral nerves, metachromatic leukodystrophy, adrenomyeloneuropathy, and giant axonal neuropathy.
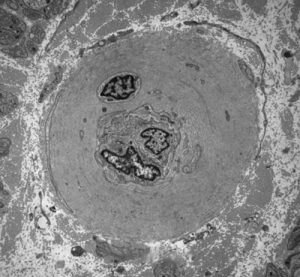
The most common cause of neuropathy in clinical practice is diabetes. Peripheral neuropathy develops in more than half of long term diabetics. Diabetes causes several types of neuropathy, which include chronic symmetrical polyneuropathy, proximal neuropathy (diabetic amyotrophy), mononeuropathies, and cranial radiculopathies. The pathogenesis of diabetic neuropathies is poorly understood. Many of them have an ischemic basis. A prominent finding in diabetic neuropathy is thickening of arterioles due to increased deposition of basement membrane material, similar to changes that occur in brain arterioles and glomerular capillaries. Nonenzymatic glycation of neural structures and other biochemical changes in diabetes probably play a role also.
Inflammatory Demyelinative Neuropathies
These uncommon neuropathies are presumed to be immune disorders in which antibodies and activated T-lymphocytes, reacting with antigens present on peripheral nerves, elicit an inflammatory and macrophage reaction that destroys myelin and axons. The strongest evidence of a humoral immune reaction in these neuropathies is that plasma exchange results in significant clinical improvement. The participation of cellular immunity is underlined by the pesence of T-lymphocytes around blood vessels in affected nerves. The two main entities in this group are the Guillain-Barré syndrome and chronic inflammatory demyelinative neuropathy. An experimental model of demyelinative neuropathy, experimental allergic neuritis (EAN), can be produced by injecting animals with myelin and Freund adjuvant or purified peripheral myelin protein P2. EAN is a cell-mediated immune reaction.
The Guillain-Barré Syndrome(GBS) is not a single disease entity. It includes several variants: Acute inflammatory demyelinative polyneuropathy (AIDP), acute motor axonal neuropathy (AMAN), and the Miller-Fisher syndrome (MFS). AIDP accounts for 90% of GBS. It begins with paresthesias in the toes and fingertips, followed by rapidly advancing weakness and areflexia. Weakness reaches a plateau within four weeks, after which recovery begins. Some cases are fulminant, evolving in one or two days. At the height of their disease, many patients are completely paralyzed and unable to breathe. Even with modern intensive care, approximately 5% of patients die from respiratory paralysis, cardiac arrest (probably due to autonomic dysfunction), sepsis, and other complications. Ten percent of those who recover have residual weakness. Though easy to diagnose in its classical form, GBS is often missed because of atypical clinical features which include ophthalmoplegia, ataxia, sensory loss, and dysautonomia. Plasma exchange (presumably removing the offending antibodies) and intravenous immunoglobulin are the treatments of choice. The two key laboratory abnormalities in GBS are decreased nerve conduction velocity or conduction block and elevated CSF protein with relatively few cells (albuminocytologic dissociation).
Peripheral nerves show perivenular mononuclear cells, demyelination (myelin proteins are the source of elevated CSF protein), and macrophages. Axonal damage, which accounts for the permanent deficits, is variable and may be severe. The pathology is most severe in spinal roots and plexuses and less pronounced in more distal nerves. In the phase of recovery, the nerve contains thin myelin sheaths, indicating myelin regeneration. AMAN shows axonal damage with little inflammation.
About 20% to 30% of GBS cases are preceded by an infection with Campylobacter Jejuni. An equal number are preceded by Cytomegalovirus (CMV) infection. The rest are preceded by Mycoplasma and other infections, or vaccinations. The bacterial wall of C. jejuni contains GM1 ganglioside. Anti-ganglioside antibodies, generated in the course of the infection, cross-react with GM1 ganglioside present in the axonal membrane at the nodes of Ranvier and in paranodal myelin. This contact elicits inflammation that damages these structures. Anti-GM1 antibodies are found in the serum of GBS patients. GBS following CMV infections has anti-GM2 antibodies.
Chronic inflammatory demyelinative polyradiculoneuropathy (CIDP) follows a chronic or relapsing course over many months or years and may cause severe permanent disability. Nerve conduction studies show decreased conduction velocity, conduction block, and prolonged distal latencies and F waves. In the active phase of the disease, the CSF shows elevated protein without increased cells. Pathologically, peripheral nerves show demyelination, thin (incompletely regenerated) myelin, and hypertrophic changes due to recurrent attacks of demyelination with intervals of repair. In chronic cases, there is significant axonal loss. Inflammation is variable, sometimes absent. The pathology is most severe in proximal nerve segments and spinal roots and may not be full blown in the sural nerve biopsy. CIDP is thought to represent an autoimmune T-cell and antibody reaction against unknown myelin antigens. Its treatment consists of plasma exchange, intravenous immunoglobulin, and corticosteroids.
The GBS and CIDP are the counterparts of MS for the peripheral nervous system. They are important, because timely intervention with plasma exchange can prevent death in the GBS and severe permanent disability in CIDP. There are standardized criteria for their diagnosis, based on the clinical, CSF, nerve conduction, and biopsy findings.
The inherited neuropathies are rare as a group and include lysosomal storage diseases, peroxisomal disorders, and familial amyloidoses. Neuropathy, in these diseases, is a component of a systemic metabolic defect. The inherited neuropathies include also a group of diseases called hereditary motor and sensory neuropathies, in which neuropathy is the main or only abnormality. The most common entity in this group and the most common overall familial neuropathy is Charcot-Marie-Tooth disease.
Charcot-Marie-Tooth disease (CMT) is not a single entity but a group of inherited neuropathies that are divided into 3 phenotypes, CMT1, CMT2, and X-linked CMT. CMT1 is the most common inherited peripheral neuropathy. It involves 1 in 2500 persons and is autosomal dominant. It causes weakness and atrophy of distal muscles, especially those innervated by the peroneal nerve (“stork leg”), pes cavus, sensory loss, and action tremor in some patients. It begins in childhood or adolescence and progresses slowly, involving other nerves. It is compatible with a normal lifespan. Nerve conduction studies show decreased conduction velocity. The nerve biopsy in CMT1 shows demyelination, myelin regeneration (thin myelin), axonal loss, and onion bulbs. In longstanding cases there is gross thickening of nerves, hence the term hypetrophic neuropathy.
CMT1 is genetically diverse. Its most common form is due to duplication of a segment of chromosome 17 (17p11.2-p12) that contains the gene for a 22 kd peripheral myelin protein, PMP22. This protein probably also plays a role in Schwann cell differentiation. CMT1 patients have three copies of the normal gene and presumably produce 1.5 times as much PMP22 as normal people do. Other forms of CMT1 are caused by mutations of the PMP22 gene or mutations of the Myelin Protein Zero (MPZ) gene. CMT2 is a distal axonopathy with a diverse genetic background. X-linked CMT is caused by mutation of a gap junction protein, connexin 32. A deletion of the PMP22 gene causes hereditary neuropathy with pressure palsies. Autosomal dominant and autosomal recessive mutations of PMP22, MPZ, and other genes cause CMT3 (Dejerine-Sottas disease), a severe infantile demyelinative hypertrophic neuropathy. These molecular abnormalities underline the importance of myelin proteins for structural stability of myelin and show how diverse genetic abnormalities can cause a similar phenotype.
Familial amyloid neuropathies (FAP) are a group of familial systemic amyloidoses with involvement of peripheral nerves. The most common FAP is caused by an autosomal dominant mutation of the transthyretin gene on 18q11. The mutant protein is deposited in the form of amyloid and damages peripheral nerves, the heart, kidneys, gastrointestinal tract, and other organs. In nerves, amyloid damages first and most severely small fibers, causing loss of pain and temperature sensation and autonomic dysfunction. Transthyretin is produced in the liver. Liver transplantation arrests the progression of the disease.
Polyarteritis nodosa and other vasculitides often involve peripheral nerves causing single or multiple mononeuropathies (due to nerve ischemia), asymmetric polyneuropathy, and distal symmetric polyneuropathy. A sural nerve biopsy along with a muscle biopsy are the best tissues for establishing the diagnosis of vasculitis. The nerve biopsy is diagnostic in over half of patients with systemic vasculitis and clinical neuropathy, and the diagnostic yield increases with the addition of a muscle biopsy. Such biopsies show necrotizing arteritis, perivascular inflammatory infiltrates, hemorrhage and hemosiderin deposition, neovascularization in epineurial arteries, and variable changes in nerve fascicles, depending on the severity and stage of neuropathy. The muscle shows vasculitis and denervation atrophy.